基因组测序解析2014年埃博拉病毒的爆发与传播
摘要:自今年二月,几内亚发现埃博拉病毒疫情后,埃博拉病毒疫情在西非多个国家爆发,在国际上引起广泛关注。为了尽快掌握此次疫情的基本情况,一个由来自四个国家五十余名研究人员组成的团队对99个埃博拉病毒基因组进行高通量测序,并推断出了关于此次疫情的来源与传播方式。
埃博拉病毒是一种能引起人类和灵长类动物产生埃博拉出血热的烈性传染病病毒,1976年在苏丹南部和刚果(金)的埃博拉河地区首次发现它的存在,“埃博拉”由此而得名。此次埃博拉病毒的爆发开始于几内亚,并随后传播至利比亚、塞拉利昂和尼日利亚。截止2014.8.19,已发现2240例病例,并导致1229人死亡。为了有效控制疫情,研究人员针对此次疫情暴发的源头及传播途径进行研究。
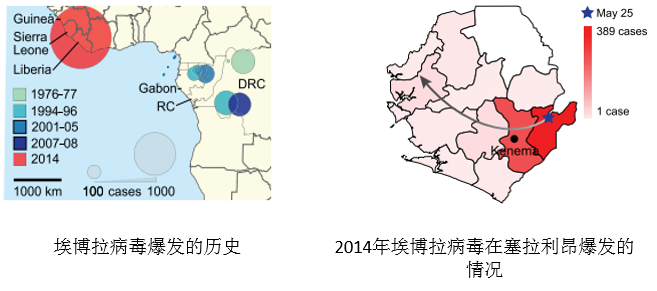
研究发现,此次埃博拉疫情在西非爆发的源头是一名前往塞拉利昂凯内马市政医院就诊的年轻女性,是塞拉利昂境内通过PCR诊断技术确诊感染埃博拉病毒的首例病例。经过调查发现,她是在一位死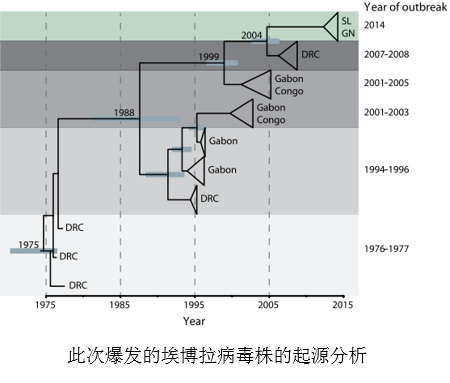 于埃博拉的医生的葬礼上感染病毒的,并且发现另外13名参加该葬礼的女性也感染了埃博拉病毒。研究人员随后从其中12名患者体内提取了15份病毒样品,进行全基因组测序。此后,研究小组又对来自塞拉利昂的66名患者的84份样品中的埃博拉病毒进行了全基因组测序(测序深度约2000x,基因组覆盖度超过99.9%),共找到341个碱基替换突变,55个单核苷酸多态性位点。研究人员通过与此前埃博拉疫情爆发时所获得的20个病毒基因组进行比较,发现此次在西非发现的埃博拉病毒都起源于同一祖先,来自于十年前在非洲中部地区爆发的那场埃博拉疫情。
于埃博拉的医生的葬礼上感染病毒的,并且发现另外13名参加该葬礼的女性也感染了埃博拉病毒。研究人员随后从其中12名患者体内提取了15份病毒样品,进行全基因组测序。此后,研究小组又对来自塞拉利昂的66名患者的84份样品中的埃博拉病毒进行了全基因组测序(测序深度约2000x,基因组覆盖度超过99.9%),共找到341个碱基替换突变,55个单核苷酸多态性位点。研究人员通过与此前埃博拉疫情爆发时所获得的20个病毒基因组进行比较,发现此次在西非发现的埃博拉病毒都起源于同一祖先,来自于十年前在非洲中部地区爆发的那场埃博拉疫情。
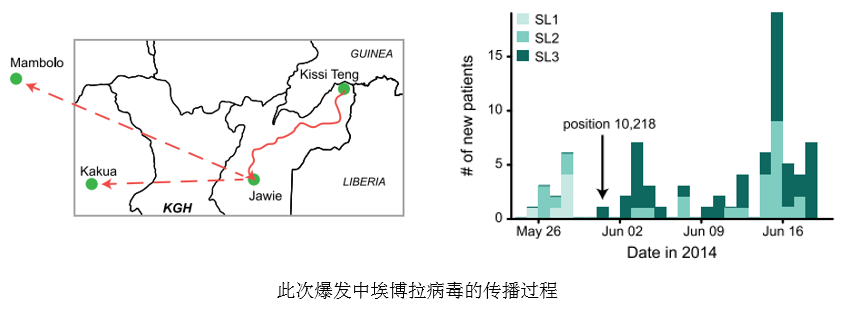
研究人员还在提取的99份病毒样品中发现了263个病毒在宿主体内寄生过程中发生的单核苷酸突变位点(intrahostsingle-nucleotide variant, iSNV)。研究人员利用这些突变信息对病毒的传播过程进行了解析。对于从首批确诊的患者体内得到的病毒样品的研究发现,这些患者分别被两株不同的埃博拉病毒所感染。根据进化分析,这两株埃博拉病毒是在今年四月份在基因组水平上发生分离的,并在五月下旬共同出现在塞拉利昂。在传播过程中,其中一株埃博拉病毒在感染一名护士后,在第10218位碱基处发生突变,产生第三株埃博拉病毒,并通过这名护士的接触者进一步蔓延,并且在之后确诊的大多数病例都是由该株病毒感染所引起的。下一步,研究人员将对埃博拉病毒基因组中新发现的突变进行深入研究,以确定突变对其感染及传播特点的影响和改变,为抗击埃博拉病毒提供更多信息。
不幸的是,有五名参与这项研究的医护人员为病人提供治疗和帮助的过程中感染埃博拉病毒,离开了人世。他们分别是舍克·汗(Sheik Humarr Khan)、穆罕默德·富拉(Mohamed Fullah)、穆巴鲁·方妮(Mbalu Fonnie)、阿列克斯·莫依格波(Alex Moigboi)和阿丽斯·科沃玛(Alice Kovoma)。
向他们致以最崇高的敬意!
参考文献:
1.Gire, S. K.and A. Goba, et al. (2014). "Genomic surveillance elucidates Ebola virusorigin and transmission during the 2014 outbreak." Science 345 (6202): 1369-72.




